软件文档:https://github.com/STAR-Fusion/STAR-Fusion/wiki
##软件安装
- STAR,https://github.com/alexdobin/STAR
- 运行依赖 perl 模块
- DB_File
- URI::Escape
- Set::IntervalTree
- Carp::Assert
- JSON::XS
- PerlIO::gzip
- common::sense
- Types::Serialiser
- Canary::Stability
##数据库准备
下载一个较小的未处理的参考文件,自己运行 index 命令。要是网速够快也可以直接在 index 好的数据库文件,~27G
1 | wget -c https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/GRCh37_v19_CTAT_lib_Feb092018.source_data.tar.gz |
##运行STAR-Fusion
STAR-Fusion 对 STAR 输出的嵌合比对分析发现可能存在的基因融合事件
从 fastq 文件开始
1 | STAR_FUSION_HOME/STAR-Fusion \ |
从 STAR 产生 Bam 文件开始
1 | STAR --genomeDir ${star_index_dir} \ |
输出文件
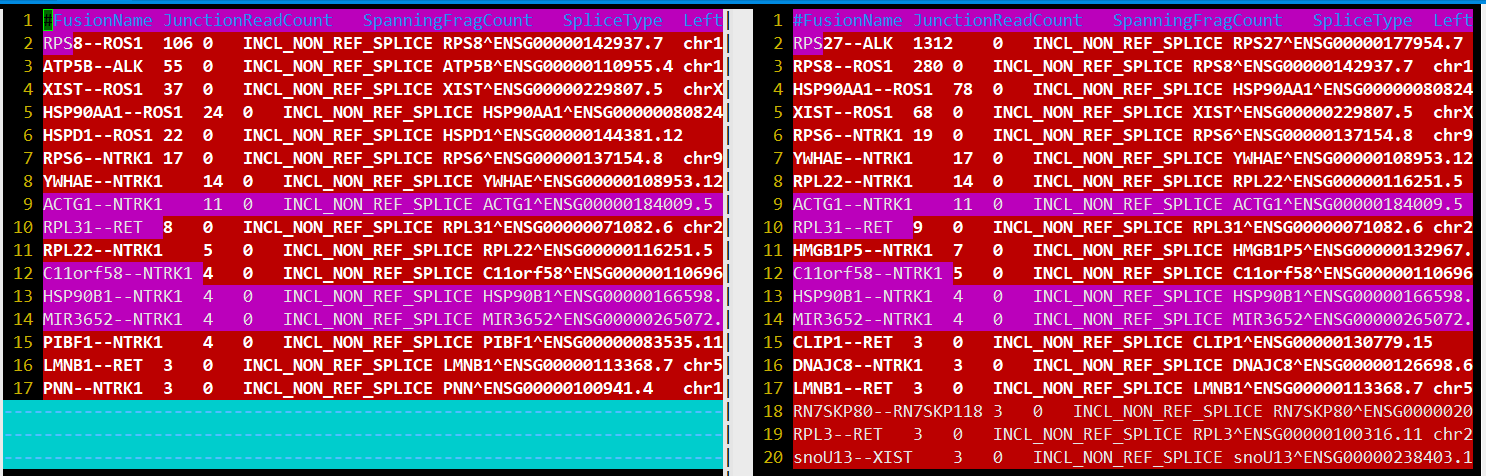
STAR 速度还是那么让人惊喜,6m reads不到半小时。 融合结果star-fusion.fusion_predictions.abridged.tsv
FusionName,
JunctionReadCount, split align到融合点的序列片段数
SpanningFragCount, 双端reads跨越融合点的序列片段数
SpliceType, 断点是否在注释文件存在
LeftGene,
LeftBreakpoint,
RightGene,
RightBreakpoint,
LargeAnchorSupport,
FFPM, fusion fragments per million total reads
LeftBreakDinuc,
LeftBreakEntropy,
RightBreakDinuc,
RightBreakEntropy,
annots,
结果比较
真是一个悲伤的故事,根据官网给的两种运行方式,结果差别这么大。查看 STAR-Fusion 脚本,使用的 mapping 参数差异有点大啊,哪一个比较合理呢(一个新坑)???