我们使用 snpEff, annovar 对得到的变异集注释,总会得到多个转录本注释结果,而只需要按照 HGVS 规范报告一个经典转录本(canonical transcripts) 对应注释结果,这时候就需要知道每个基因对应的经典转录本。
一般情况下,我们使用基因对应的最长转录本代表该基因,具体操作上,首先从 HGNC gene_with_protein_product.txt 下载经过确认的基因对应经典转录本信息,其中不确定基因再使用最长转录本代表。
HGNC 下载编码蛋白基因对应转录本
1
wget -c ftp://ftp.ebi.ac.uk/pub/databases/genenames/new/tsv/locus_types/gene_with_protein_product.txt
根据表格,很方便得到基因名字对应 hgnc_id, entrez_id, ensembl_gene_id, refseq_accession 等多数据库基因ID,使用 refseq_accession 进行 HGVS 表示,这里就只关注 refseq_accession , 还是可以发现其中一些基因名并不能对应到唯一 refseq_accession,下面就需要我们进行第二步,结合 GFF 文件挑出经典转录本。
NCBI下载人类基因注释GFF文件
1
wget -c ftp://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh37_latest/refseq_identifiers/GRCh37_latest_genomic.gff.gz
使用脚本将 gff 转换为表格样式,方便筛选每个基因对应最长转录本,具体操作参考如下,其中 pyGTF 为用来解析 gff 文件。
输出文件可以下载 hg37.geneinfo.txt , 其具体格式为:
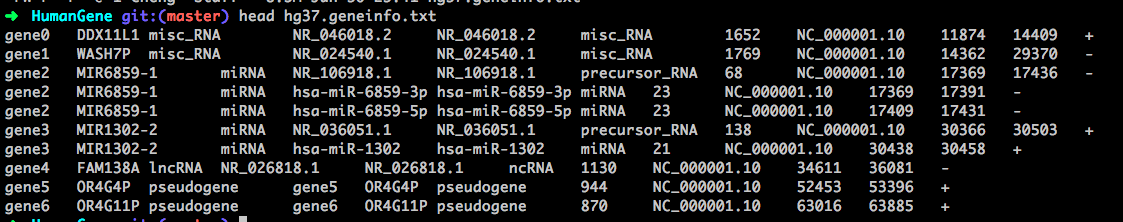
表头分别是:gene_id, gene_name, gene_type, transcript_id, transcript_name, transcript_type, transcript_length, chromosome, start, end, strand,根据 gene_name, transcript_name, transcript_length 三列排序,就可以很方便筛选出基因对应最长转录本。
脚本整合 HGNC 和 GFF 两部分结果
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30import pandas as pd
gene, length = {}, {}
gff = pd.read_table('hg37.geneinfo.txt', header=None)
gff = gff[(gff[2]=='protein_coding') | (gff[5]=='protein_coding')]
nrow, ncol = gff.shape
for index in range(nrow):
transcriptid = gff.iloc[index,3]
try:
transcriptid = transcriptid[:transcriptid.rindex('.')]
except:
pass
gene.setdefault(gff.iloc[index,1], []).append(transcriptid)
length[transcriptid] = gff.iloc[index,6]
hgnc = pd.read_table('gene_with_protein_product.txt')
hgnc['symbol'] = hgnc['symbol'].astype(str)
hgnc['refseq_accession'] = hgnc['refseq_accession'].astype(str)
nrow, ncol = hgnc.shape
for index in range(nrow):
genename = hgnc.loc[index, 'symbol']
refseq = hgnc.loc[index, 'refseq_accession']
tmp = refseq.split('|')
if len(tmp) == 1:
print('{}\t{}\thgnc'.format(genename, refseq))
else:
if len(tmp) == 0:
tmp = gene[genename]
tmp = sorted(tmp, key=lambda x: length.get(x, 0), reverse=True)
print('{}\t{}\tgff'.format(genename, tmp[0]))