Exomiser summary

Limitation
- 仅能分析基因 coding region 和 exon-intron 剪接点附近变异
prioritization
PHIVE, PhenoDigm algorithm
支持数据
- 小鼠 phenotype data from MGD, IMPC
- HPO
####算法
Ontologies
Pairwise alignment of ontology concepts with OWLSim
$$ sim_j(p,q) = |\alpha^p \cap \alpha^q| / |\alpha^p \cup \alpha^q| $$
Jaccard Index
$$ IC_{(concept)} = -log2(item_{concept}/item) $$
IC is calculated for the Least Common Subsuming (LCS) phenotype of the pair of concepts
Determining phenotype similarity score estimation
$$ maxScore(Q, D) = max(score(i, j)) $$
i = 1 … m for Query, j = 1 … n for Disease
$$ avgScore(Q, D) = \frac {\displaystyle \sum_{i=1}^m max(score(i, j), j=1…n) + \displaystyle \sum_{j=1}^n max(score(i, j), i=1…m)}{m + n} $$
Dvaluated the performance of recalling known disease–gene or model–disease associations use the
combinedPercentageScore- $$ maxPercentageScore(a, b) = \frac {maxScore(a, b)} {maxScore(a, optimal match of a)} $$
- $$ avgPercentageScore(a, b) = \frac {avgScore(a, b)} {avgScore(a, optimal match of a)} $$
- $$ combinedPercentageScore = avg(maxPercentageScore(a, b), avgPercentageScore(a, b)) $$
####限制
- 突变基因须存在对应的小鼠模型
- 需要整合模式动物 MP 和 HPO,生成包含 MP 和 HPO 对应信息的 Ontologies,该步骤用到词义匹配
http://purl.obolibrary.org/obo/uberon/basic.obo
PhenIX, Phenomizer algorithm
支持数据
HPO 数据库中与 OMIM 数据库关联数据
预计算每个 HPO 项对应的 IC(Information Content)
计算每个 HPO term 在数据库中关联的 4813 中遗传病中出现的频率,然后去负对数得到每个 term 的 IC
算法
Information content: IC
the negative natural logarithm of the frequency
similarity: MICA
The similarity between two terms can be calculated as the IC of their most informative common ancestor (MICA)
HPO input as Query
$$ sim(Q \rightarrow D) = avg[\displaystyle \sum_{q1 \in Q}\ max_{d1 \in D}(IC(MICA(q1, d1)))] $$
symmetric similarity
$$ sim_{symmetric}(Q,D) = (sim(Q\rightarrow D) + sim(D\rightarrow Q)/2) $$
p Value Calculation
The p values are estimated by Monte Carlo random sampling and corrected for multiple testing by the method of Benjamini and Hochberg
Query term 超过 10 个时候,为了减少计算量,全部 downsample 到 10 个 query 计算 pvalue;
基于以上逻辑,该部分工作为完全重复性工作,预先计算完成。
限制
- 仅能分析已知的孟德尔疾病
ExomeWalker
支持数据
- 需要输入临床疾病对应的已知致病基因
- protein-protein interaction data from STRING(score >= 0.7)
算法
PPI, protein-protein interaction network
使用 STRING 数据库中 score >= 0.7 的互作关系
Disease-gene families,导致相似临床表型的一系列基因,分析中由用户输入
Variant score
- 根据基因频率添加一个 0-1 得分,分别对应基因频率于 2%-0%,MAF>2% 对应 0。1% 以上的 variant 被丢弃
- 预测的致病性结果,计算一个 致病性得分 被计算
- 位于非编码区域和非剪接点的 脱靶变异 得分为0,同时被丢弃;
- 非同义突变得分为 SIFT 或 MutationTaster 预测得分;
- gene 和疾病的关系从 OMIM 数据库中提取;
- 上述三种方式都未提及的 variant,致病性得分被指定为 0.6.
- 最终变异优先级评分由 频率得分 * 致病性得分 得到
Gene score, Random walk analysis
弃疗 ……
ExomeWalker score
combination of the random walk score and the best scoring variant in that gene
AR inheritance, variant score of gene = avg(top 2 of variant score)
Logistic regression on a training set of 20000 disease variants and 20 000 benign variants
10-fold cross validation was used to train and test the model
This final ExomeWalker score gives a measure from 0 to 1
限制
- 需输入疾病已知致病基因
- 通常针对异质性较高的疾病,用于发现新的致病基因位点
hiPHIVE
使用 PhenoDigm 算法分析斑马鱼突变体表型数据,与上述三种方法结果进行整合,最总结果用于 vairant rank
Information content
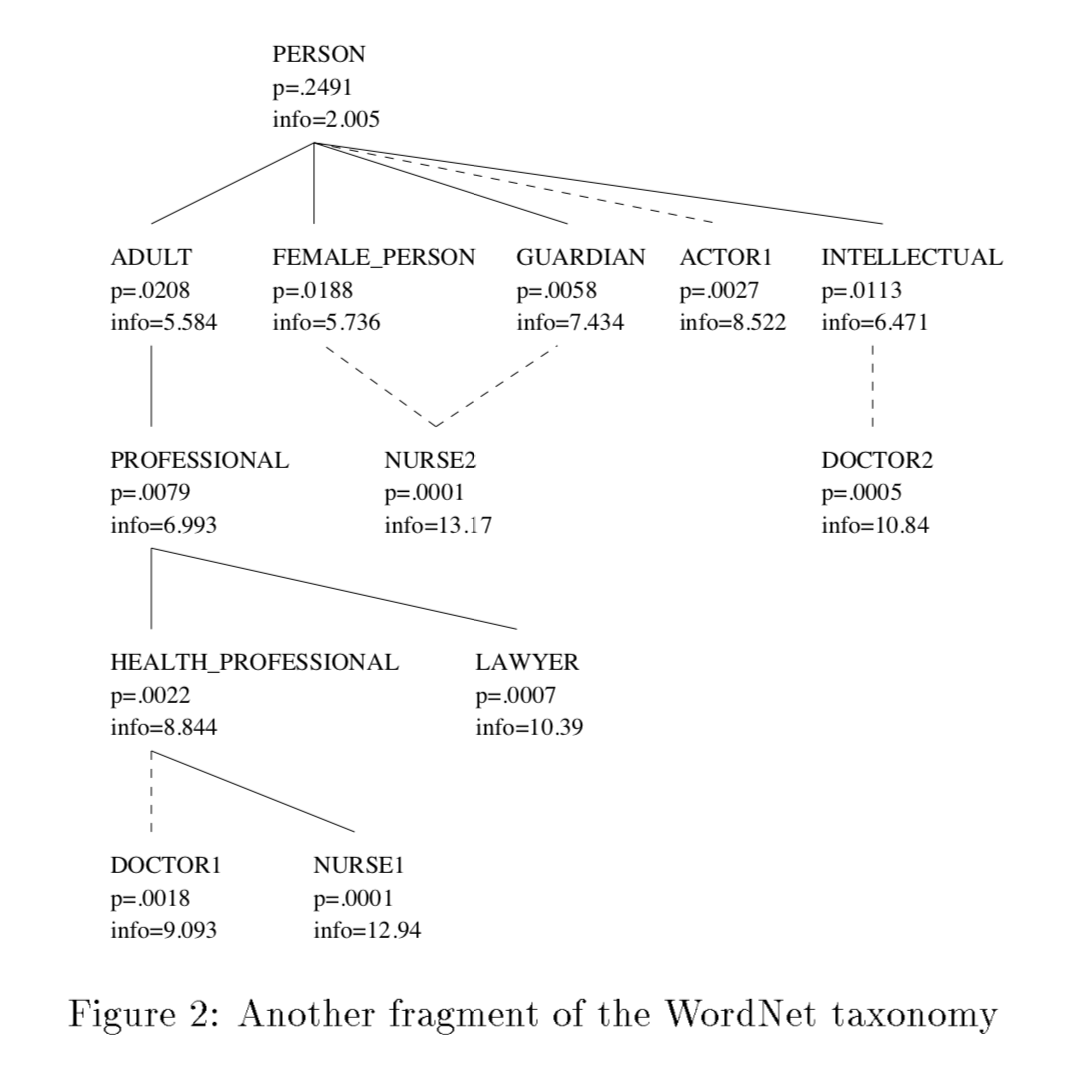
IC( information content ) 原始定义文献参考 10.1613/jair.514,首先构建如图 Ontologies ,根据出现频率负对数的定义计算每一项对应的 IC,其中 DOCTOR1 和 NURSE1 分别对应的 IC 是 9.093, 12.94,两者的相似性定义为最近共同祖先的 IC,即为 8.844。
IC 算法应用到人类疾病中,对临床表型和已知疾病表型进行相似性计算,详细逻辑原理体现如图:

文献
- Smedley, D., Jacobsen, J. O. B., Jäger, M., Köhler, S., Holtgrewe, M., Schubach, M., … Robinson, P. N. (2015). Next-generation diagnostics and disease-gene discovery with the Exomiser. Nature Protocols, 10(12), 2004–2015. https://doi.org/10.1038/nprot.2015.124
- Smedley, D., Oellrich, A., Köhler, S., Ruef, B., Westerfield, M., Robinson, P., … Mungall, C. (2013). PhenoDigm: Analyzing curated annotations to associate animal models with human diseases. Database. https://doi.org/10.1093/database/bat025
- Köhler, S., Schulz, M. H., Krawitz, P., Bauer, S., Dölken, S., Ott, C. E., … Robinson, P. N. (2009). Clinical Diagnostics in Human Genetics with Semantic Similarity Searches in Ontologies. American Journal of Human Genetics, 85(4), 457–464. https://doi.org/10.1016/j.ajhg.2009.09.003
- Resnik, P. (1999). Semantic Similarity in a Taxonomy: An Information-Based Measure and its Application to Problems of Ambiguity in Natural Language. Journal of Artificial Intelligence Research, 11, 95–130. https://doi.org/10.1613/jair.514
- Smedley, D., Köhler, S., Czeschik, J. C., Amberger, J., Bocchini, C., Hamosh, A., … Robinson, P. N. (2014). Walking the interactome for candidate prioritization in exome sequencing studies of Mendelian diseases. Bioinformatics. https://doi.org/10.1093/bioinformatics/btu508