1/1/2018
根据与基因的相对位置,lncRNA可以分为Intergenic LncRNAs(lincRNA), Bidirectional LncRNAs, Intronic LncRNAs, Antisense LncRNAs, Sense-overlapping LncRNAs五类,每类详细定义规则如下:
定义
ref: https://www.arraystar.com/reviews/v30-lncrna-classification/
Intergenic LncRNAs
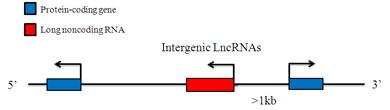
Intergenic LncRNAs are long non-coding RNAs which locate between annotated protein-coding genes and are at least 1 kb away from the nearest protein-coding genes. They are named according to their 3-protein-coding genes nearby. Gene expression patterns have implicated these LincRNAs in diverse biological processes, including cell-cycle regulation, immune surveillance and embryonic stem cell pluripotency. LincRNAs collaborate with chromatin modifying protein (PRC2, CoREST and SCMX) to regulate gene expression at specific loci.
Bidirectional LncRNAs
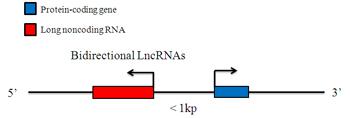
A Bidirectional LncRNA is oriented head to head with a protein-coding gene within 1kb. A Bidirectional LncRNA transcript exhibits a similar expression pattern to its protein-coding counterpart which suggests that they may be subject to share regulatory pressures. However, the discordant expression relationships between bidirectional LncRNAs and protein coding gene pairs have also been found, challenging the assertion that LncRNA transcription occurs solely to “open” chromatin to promote the expression of neighboring coding genes.
Intronic LncRNAs
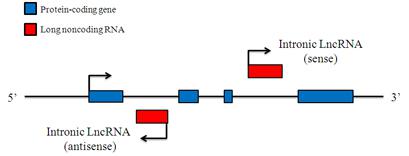
Intronic LncRNAs are RNA molecules that overlap with the intron of annotated coding genes in either sense or antisense orientation. Most of the Intronic LncRNAs have the same tissue expression patterns as the corresponding coding genes, and may stabilize protein-coding transcripts or regulate their alternative splicing.
Antisense LncRNAs
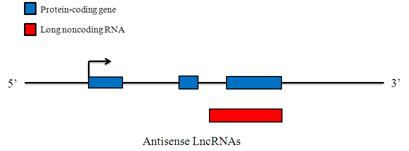
Antisense LncRNAs are RNA molecules that are transcribed from the antisense strand and overlap in part with well-defined spliced sense or intronless sense RNAs. Antisense-overlapping LncRNAs have a tendency to undergo fewer splicing events and typically show lower abundance than sense transcripts. The basal expression levels of antisense-overlapping LncRNAs and sense mRNAs in different tissues and cell lines can be either positively or negatively regulated. Antisense-overlapping LncRNAs are frequently functional and use diverse transcriptional and post-transcriptional gene regulatory mechanisms to carry out a wide variety of biological roles.
Sense-overlapping LncRNAs
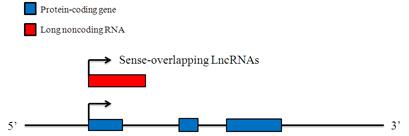
These LncRNAs can be considered transcript variants of protein-coding mRNAs, as they overlap with a known annotated gene on the same genomic strand. The majority of these LncRNAs lack substantial open reading frames (ORFs) for protein translation, while others contain an open reading frame that shares the same start codon as a protein-coding transcript for that gene, but unlikely encode a protein for several reasons, including non-sense mediated decay (NMD) issues that limits the translation of mRNAs with premature termination stop codons and trigger NMD-mediated destruction of the mRNA, or an upstream alternative open reading frame which inhibits the translation of the predicted ORF.
实现
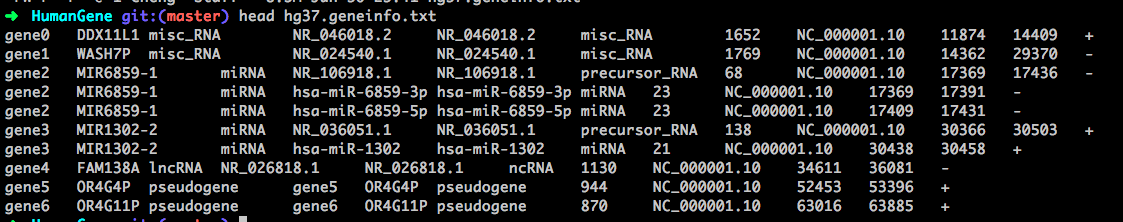
1 | step0 prepare |