annovar可以说是最常用的变异注释软件了,除了基于基因位置进行注释,还有丰富的第三方数据库支持,clinvar, cosmic等等,但是annovar提供下载数据库版本较老,需要自行下载第三方 vcf 进行转换。
以 clinvar 为例说说 vcf 格式转换成 annovar 可识别表格格式,annovar 仅需要 vcf 中部分信息,为了脚本通用这里输出vcf所有信息到 tab 格式,详细处理脚本如下,同时实现脚本存放 github。
1 | import gzip |
annovar可以说是最常用的变异注释软件了,除了基于基因位置进行注释,还有丰富的第三方数据库支持,clinvar, cosmic等等,但是annovar提供下载数据库版本较老,需要自行下载第三方 vcf 进行转换。
以 clinvar 为例说说 vcf 格式转换成 annovar 可识别表格格式,annovar 仅需要 vcf 中部分信息,为了脚本通用这里输出vcf所有信息到 tab 格式,详细处理脚本如下,同时实现脚本存放 github。
1 | import gzip |
编写分析流程时候,为了兼容性总会设置一些常用选项配置文件输入,不同配置方式各有一些特点,就用过的几种配置方式简单整理
1 | import os |
书写方式完全按照python脚本进行,可读性较高,问题在于PYTHONPATH的处理,脚本环境内键值对可能产生的覆盖问题,准确导入分析流程
1 | import configparser # python3 |
但是,在configparser模块中不区分section, option大小写,一次读取一条配置信息
1 | import json |
json文件格式要求较严格,书写配置文件容易手误出错,相比之下更适用于流程中数据传递
1 | import yaml |
1 | # 表示 key-value 对 |
相比于上面几种方法,yaml可读性高,容易书写,不用考虑PYTHONPATH问题,批量导入配置信息
http://varnomen.hgvs.org/recommendations/, 描述规则基本格式为:[identifier]:[levels].[Description of variant]

其他具体要求描述可以参考:http://varnomen.hgvs.org/bg-material/refseq/
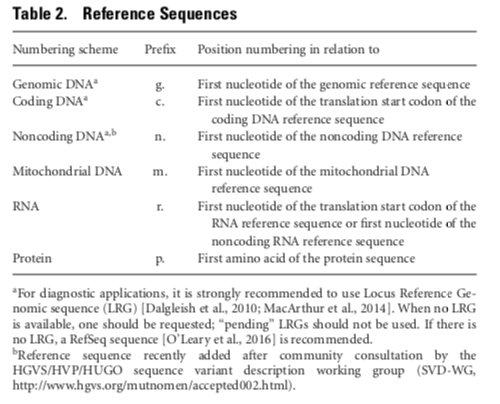
Reference Sequence Types主要有g, c, n, m, r, p六种,其中c, p, g使用最多
c.255C>Ap.Trp24Cys, p.Trp24*, p.Cys188=c.19_21delp.Lys23_Val25delc.240_241insAGGp.Lys2_Gly3insGlnSerLysc.20_23dupp.Ser6dupc.142_144delinsTGGp.Arg48Trpp.Arg97fs
g.123A>G 和 p.Trp26Terpandas 表格数据处理,唰唰唰…
1 | import pandas as pd |
1 | import pandas as pd |
1 | import pandas as pd |
argsparse 解析命令行参数,一些常用参数整理。
1 | import argparse |
python 脚本内执行 linux 命令有多种方式,os.system(), os.popen()也可以,官方最新推荐方式为subprocess
1 | import subprocess |
os 模块实现 linux cmd 执行
1 | import os |
tophat2-fusion 文档搬运工:https://ccb.jhu.edu/software/tophat/fusion_tutorial.shtml
###step1.tophat
1 | tophat2 -p 8 -o $OUTPUT --fusion-search --keep-fasta-order --bowtie1 --no-coverage-search --fusion-anchor-length 10 --fusion-min-dist 100000 --mate-inner-dist 40 --mate-std-dev 100 $hg_bowtie1_index $read1.fastq.gz $read2.fastq.gz |
Note:
–fusion-search,融合 reads 搜索
–keep-fasta-order,
–bowtie1,使用 bowtie1 代替 bowtie2 进行 reads 比对。需要使用 bowtie-1.1.2,最新版本 1.2.2 会报错
–no-coverage-search,
–fusion-anchor-length 10 ,
–fusion-min-dist 100000 ,
–mate-inner-dist 40 ,
–mate-std-dev 100 ,
###step2.tophat-fusion-post
1 | tophat-fusion-post -p 8 --num-fusion-reads 1 --num-fusion-pairs 2 --num-fusion-both 5 $hg_bowtie1_index |
Note:
–num-fusion-reads* , 支持融合的最少 reads 数目
–num-fusion-pairs, 支持融合事件的最少配对 reads 数,表示为跨越融合点的测序片段
–num-fusion-both, 支持融合的最少 reads,包括跨越的 reads 对和 split reads
–fusion-read-mismatches, Reads support fusions if they map across fusion with at most this many mismatches. The default is 2.
–fusion-multireads, Reads that map to more than this many places will be ignored. The default is 2.
–non-human, If your annotation is different from that of human, use the option.
–fusion-pair-dist, Pairs with this specified distance are counted as supporting evidence for the fusion. The default threshold for the inner distance is 250.
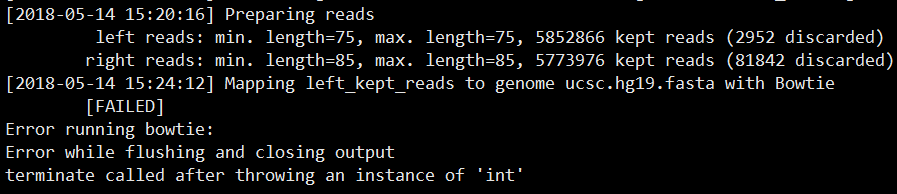
测试了多个样品,多个参数,也根据官网数据、命令运行项目,都没有发现任何融合结果,bug还是打开方式不对
软件文档:https://github.com/STAR-Fusion/STAR-Fusion/wiki
##软件安装
##数据库准备
下载一个较小的未处理的参考文件,自己运行 index 命令。要是网速够快也可以直接在 index 好的数据库文件,~27G
1 | wget -c https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/GRCh37_v19_CTAT_lib_Feb092018.source_data.tar.gz |
##运行STAR-Fusion
STAR-Fusion 对 STAR 输出的嵌合比对分析发现可能存在的基因融合事件
1 | STAR_FUSION_HOME/STAR-Fusion \ |
1 | STAR --genomeDir ${star_index_dir} \ |
STAR 速度还是那么让人惊喜,6m reads不到半小时。 融合结果star-fusion.fusion_predictions.abridged.tsv
FusionName,
JunctionReadCount, split align到融合点的序列片段数
SpanningFragCount, 双端reads跨越融合点的序列片段数
SpliceType, 断点是否在注释文件存在
LeftGene,
LeftBreakpoint,
RightGene,
RightBreakpoint,
LargeAnchorSupport,
FFPM, fusion fragments per million total reads
LeftBreakDinuc,
LeftBreakEntropy,
RightBreakDinuc,
RightBreakEntropy,
annots,
真是一个悲伤的故事,根据官网给的两种运行方式,结果差别这么大。查看 STAR-Fusion 脚本,使用的 mapping 参数差异有点大啊,哪一个比较合理呢(一个新坑)???
trimmomatic 使用 java 编写,免安装多平台运行,同时运行速度非常快。
1 | paired end |
参数选择
PE/SE
设定对 Paired-End 或 Single-End 的 reads 进行处理,其输入和输出参数稍有不一样
threads
设置多线程运行数
phred33/phred64
设置碱基的质量格式,可选 phred64
ILLUMINACLIP:
切除 adapter 序列。参数后分别接 adapter 序列的 fasta 文件,允许的最大 mismatch 数, palindrome 模式下匹配碱基数阈值: simple 模式下的匹配碱基数阈值
minAdapterLength:只对 PE 测序的 palindrome clip 模式有效,指定 palindrome 模式下可以切除的接头序列最短长度,由于历史的原因,默认值是 8,但实际上 palindrome 模式可以切除短至 1bp 的接头污染,所以可以设置为 1 。
keepBothReads:只对 PE 测序的 palindrome clip 模式有效,这个参数很重要,在上图中 D 模式下, R1 和 R2 在去除了接头序列之后剩余的部分是完全反向互补的,默认参数 false,意味着整条去除与 R1 完全反向互补的 R2,当做重复去除掉,但在有些情况下,例如需要用到 paired reads 的 bowtie2 流程,就要将这个参数改为 true,否则会损失一部分 paired reads。
SLIDINGWINDOW
从 reads 首端( 5’端)开始进行滑动,当滑动位点周围一段序列(window)的平均碱基低于阈值,则从该处进行切除。 Windows 的 size 是 4 bp, 若其平均碱基质量小于15,则切除
MAXINFO:
LEADING/TRAILING>
切除 reads 首端( 5’端) / reads 末端( 3’端)碱基质量小于指定值的碱基
CROP/HEADCROP
从 reads 末端( 3’端)/reads 首端( 5’端)切除碱基到指定长度
MINLEN
抛弃低于指定长度的 reads
TOPHRED33/TOPHRED64
转换碱基质量格式,Illumina HiSeq 2000质量系统为phred-64,可用该参数转换到phred-33
日常工作之读文档,了解参数改变、新方法功能,对已有流程进行修改。
各种文档看一边遍一遍,使用需要又一次看 SnpEff (http://snpeff.sourceforge.net/index.html ),进行一些整理如下,不过还是建议原文,鬼知道我会在哪里写错理解错。
annotation
1 | java -jar snpEff.jar eff genome input.vcf >output.vcf |
database
人、小鼠等物种都有已经 build 的数据库,可以直接下载,链接:https://sourceforge.net/projects/snpeff/files/databases/v4_3/,注释选择基因组版本。
1 | java -jar snpEff.jar databases # 查看可用数据库 |
软件还提供了自行构建数据库,详细用法如下:
1 | 添加配置信息 |
此外,snpEff 构建数据库还支持 RefSeq, GenBank 等多种数据输入格式,具体用法查看原文档。
注意物种 codon 选择,codon 可以在 https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi 中查看,codon可以在~/snpEff/snpEff.config文件中设置:
1 | # 设置特异 codon |
dump
从数据库中提取出注释信息
1 | java -jar snpEff.jar dump -v -bed GRCh37.70 > GRCh37.70.bed |
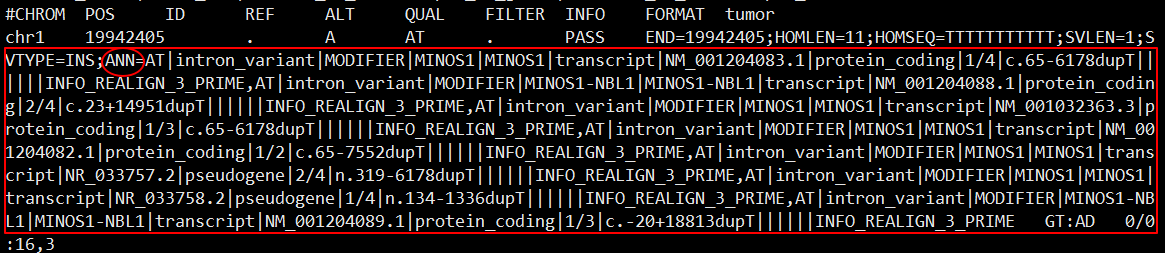
可以看到注释信息被添加到了 VCF 中每个变异 INFO 信息中,以 ANN= 特征开始,详细的注释说明可查看官方文档: http://snpeff.sourceforge.net/VCFannotationformat_v1.0.pdf。因为基因多个转录本、相互重叠基因等原因,可以看到变异位点被多次注释,需根据实际需求进行筛选。