df -h查看卸载硬盘信息umount /dev/sdf,停止使用的硬盘假装忙碌状态1
2
3umount: /mnt/usbhd2: device is busy.
(In some cases useful info about processes that use
the device is found by lsof(8) or fuser(1))fuser /dev/sdf,查看占用硬盘的进程1
/mnt/sdf: 106657c
kill -9 106657重新执行
umount /dev/sdf
shell 命令使用笔记
&&,cmd1 && cmd2如果cmd1成功执行(返回0,注意设置中脚本执行返回值),那么执行cmd2
||,cmd1 || cmd2如果cmd1执行失败,那么执行cmd2
(),{}组合逻辑控制()组合来控制命令; {}组合控制子shell
条件判断
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26if [ condition ]; then
if test condition; then
echo 'yes'
fi
使用[]判断条件时,[]和condition之间必须用空格分隔开
-d 判断是不是文件夹
-f 判断是不是文件
-L 判断是不是链接
-s 判断文件是否存在且非空
-r 判断文件是否可读
-w 判断文件是否可写
-x 判断文件是否可执行
-z 判断是否空字符串
-n 判断是否非空字符串
= 判断字符串是否相等
!= 判断字符串是否不等
-eq 判断数值是否相等
-ne 判断数值是否不等
-gt 第一个数大于第二个数
-ge 第一个数大于等于第二个数
-lt 第一个数小于第二个数
-le 第一个数小于等于第二个数
-a 逻辑与
-o 逻辑或
! 逻辑否退出状态
exit n,其中n表示数字,0表示脚本执行成功无错误,1表示执行失败某处有错误可以使用 $? 获得最后执行命令推出状态
shell 脚本模版
1
2
3
4
5!/usr/bin/env bash # 指定 shell 执行程序
!/bin/bash # option B
set -x # 打印出正在执行的 cmd
set -e # shell 中任一 cmd 执行推出状态不为 0 时,直接退出整个 shell 任务set -e针对多 cmd ,且存在依赖关系的 shell 运行非常有用shell脚本
[ ] $# 获取脚本输出参数长度
[ ] $@ 获取脚本所有参数
循环
1
2
3
4for i in `ls `
do
echo 'yes'
done序列生成
1
2
3seq 3 # 1 2 3
seq 3 5 # 3 4 5
seq 5 2 10 # 5 7 9随机数生成
1
2
3
4
5
6
7echo $RANDOM
RANDOM是bash的一个内建函数,会返回一个[0, 32676]内的整数
生成一定范围内的随机数
beg=5
end=15
echo $((RANDOM % ($end - $beg) + $beg))shell 数学计算
1
2
3echo $((9-4)) # 5
echo $((9/4)) # 2
echo $((9/-4)) # -2多列参数对文件进行排序
1
sort -k1,1 -k2,2n sort_demo.txt # 一定是 -k1,1 形式
解包
1
2
3
4
5
6
7
8
9head -n 5 hg19-tRNAs_map.txt
chr1.trna1 tRNA-Val-CAC-13-1
chr1.trna10 tRNA-Asn-GTT-16-1
chr1.trna100 tRNA-Asn-GTT-6-1
cat hg19-tRNAs_name_map.txt | while read pos name; do echo "Position: $pos Name: $name"; done | head -n 5
Position: chr1.trna1 Name: tRNA-Val-CAC-13-1
Position: chr1.trna10 Name: tRNA-Asn-GTT-16-1
Position: chr1.trna100 Name: tRNA-Asn-GTT-6-1
提交测序数据到 SRA
九月一号开学了,暑假作业写完了没有?但是跟我并没有关系,不开学,没作业,但是好多事情啊,,,最近提交了一些数据到SRA,每次过程总是那么曲折离奇,对一些遇到的问题进行整理填坑吧。
简单说,申请BioProject,申请BioSample,填写SRA_metadata,上传数据,虽然过程清晰,坑还没填完,仍在邮件NCBI,过程供参考。
1. BioProject
申请一个项目编号,填写submitter 信息,选择项目类型(Raw sequence reads),物种名称,释放时间,项目描述,BioSample和文章信息(跳过,跳过,,),核对提交
2. BioSample
申请一个样品编号,submitter 信息,释放时间,样品类型,填写样品属性表格,核对提交
3. SRA
主要就填写SRA_metadata,测序平台,文库类型等等,选择批量提交,填写表格注意看要求啊,除了样品名称、登录号等信息,其他属性组合每行也要是唯一的。
4. submit data
看数据大小,选择合适上传方法吧,小于2G用aspera浏览器插件,大数据选择ftp或者aspera命令行。这几次都用了aspera cmd上传,霸占几乎全部网速,很快上传好
具体命令为:ascp -i <path/to/key_file> -QT -l100m -k1 -d <path/to/folder/containing files> subasp@upload.ncbi.nlm.nih.gov:uploads/XXX@xxx.com_g3O1FgOE, 其中key必须是全路径,-d接包括原始数据的文件夹(不再包含其他文件夹),XXX@xxx.com为注册账号邮箱
上传结束后,SRA中选择上传数据文件夹,,,
好了,装逼结束,文件没仔细检查,传了两不完整的fastq,发邮件给ncbi沟通呢,md5值也还没有用到,不知什么情况,,,too young, too naive…
python class 实现 fasta 文件解析
没有对象怎么办(object)???当然是new一个啊
1 | class Sequence(object): |
懒癌发作了,就这,,,
服务器挂载 4T 硬盘
因为一些我并不理解的原因,服务器不能正确识别4T的NTFS移动硬盘,需要对其格式化成ext4格式,就我的一些操作记录。
fdisk -l,查看硬盘分配的硬件编号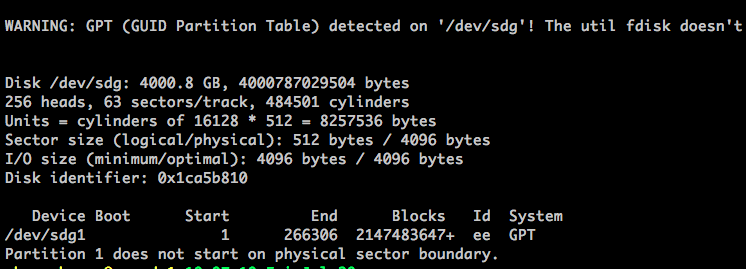
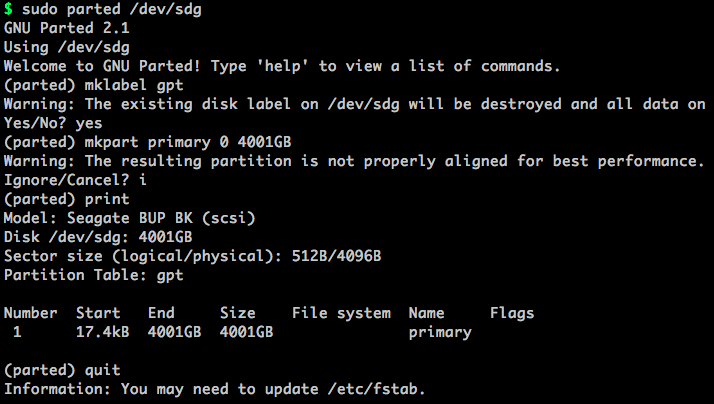
parted /dev/sdg,对使用parted工具对硬盘进行分区mklable gpt,把磁盘格式化为 gpt分区表mkpart primary 0 4000G,创建一个主分区,起始位置-结束位置mkpart extended 2000G 4000G,可选,创建一个扩展分区print,打印出当前设备的信息quit,推出parted
mkfs.ext4 /dev/sdg1,对硬盘进行格式化,如果存在多个分区,每个分区都需要进行格式化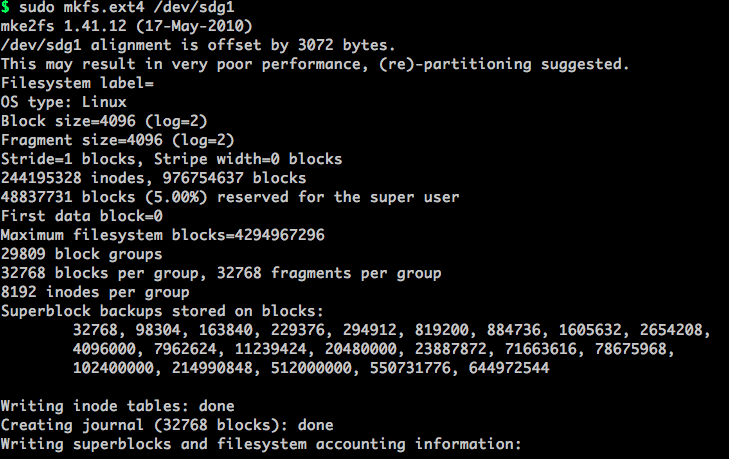
mount /dev/sdg1 /mnt/usbmount1,挂载硬盘到服务器umount /dev/sdg1,硬盘卸载
一次诡异的 python 错误排查
输入表格示例:
| library | reads_num | reads_len | bases_num | GC | avgQ | Q20 | Q30 |
|---|---|---|---|---|---|---|---|
| A-Ctrl | 46,875,170 | 150.0 | 7.00 Gb | 49.90% | 36.6 | 93.24% | 85.97% |
| B-Ctrl | 154,351,882 | 150.0 | 23.00 Gb | 45.40% | 36.9 | 93.80% | 86.93% |
| C-Ctrl | 58,126,912 | 150.0 | 8.00 Gb | 60.05% | 36.05 | 92.19% | 83.96% |
| D-Ctrl | 143,761,494 | 150.0 | 21.00 Gb | 42.00% | 37.25 | 94.46% | 88.15% |
初次尝试
1 | # 错误的结果 |
结果输出
[['A',
'46,875,170',
'150.0',
'7.00 Gb',
'49.90%',
'36.6',
'93.24%',
'85.97%',
'B',
'154,351,882',
'150.0',
'23.00 Gb',
'45.40%',
'36.9',
'93.80%',
'86.93%',
'C',
'58,126,912',
'150.0',
'8.00 Gb',
'60.05%',
'36.05',
'92.19%',
'83.96%',
'D',
'143,761,494',
'150.0',
'21.00 Gb',
'42.00%',
'37.25',
'94.46%',
'88.15%'],
['A',
'46,875,170',
'150.0',
'7.00 Gb',
'49.90%',
'36.6',
'93.24%',
'85.97%',
'B',
'154,351,882',
'150.0',
'23.00 Gb',
'45.40%',
'36.9',
'93.80%',
'86.93%',
'C',
'58,126,912',
'150.0',
'8.00 Gb',
'60.05%',
'36.05',
'92.19%',
'83.96%',
'D',
'143,761,494',
'150.0',
'21.00 Gb',
'42.00%',
'37.25',
'94.46%',
'88.15%'],
...]
正确解析
1 | # 正确代码 |
结果输出
[['library', 'A', 'B', 'C', 'D'],
['reads_num', '46,875,170', '154,351,882', '58,126,912', '143,761,494'],
['reads_len', '150.0', '150.0', '150.0', '150.0'],
['bases_num', '7.00 Gb', '23.00 Gb', '8.00 Gb', '21.00 Gb'],
['GC', '49.90%', '45.40%', '60.05%', '42.00%'],
['avgQ', '36.6', '36.9', '36.05', '37.25'],
['Q20', '93.24%', '93.80%', '92.19%', '94.46%'],
['Q30', '85.97%', '86.93%', '83.96%', '88.15%']]
原因分析
1 | # 错误原因查找 |
error method...
4435237272
4435237272
4435237272
4435237272
4435237272
4435237272
4435237272
4435237272
right method...
4435236984
4435354904
4435318688
4435237416
4435318040
4435319768
4435318472
4435319624
python独特的变量命名方式:变量名(>= 1个)指向储存数据物理地址,使用其中任何一个名称都可以对数据进行操作
python 字典
字典也是哈希表,键(key)必须是唯一的可哈希对象,但值(value)则不必。python中键值间使用“:”分割,不同键值对间使用“,”分割,整个字典包括在“{}”中。
1 | #初始化定义 |
设置字典默认值
1 | #key不存在时,返回自定义value |
树结构
1
2
3
4
5
6
7from collections import defaultdict
def tree(): return defaultdict(tree)
users = tree()
users['codingpy']['username'] = 'earlgrey'
#甚至还能不赋值
users['Python']['Standard Library']['os']
计算list中某元素出现次数
1 | # 方法一 |
Linux 命令 —— find
find,Linux常用文件搜索命令简要整理
基本用法
find基本用法如:find [path] <search regular> [action]
- 可选参数path,支持空格分隔的多路径查找,默认为当前路径
- 可选参数action
- -print,匹配文件输出到stdout,默认
- -exec,对find匹配到的文件执行该参数所给出的shell命令,格式为command { } \;,其中{}表示匹配文件
- -ok,于-exec类似,执行前给出提示是否执行
文件名
1
2
3find [path] -name <name>
find [path] -iname <name> #不区分大小写
其中<name>支持通配符,一般使用时建议增加单引号文件类型,一般配合其他搜索参数一起使用
1
2find [path] -type [bdcplf]
其中d表示目录,l表示链接,f表示文件文件大小
1
2
3-size 参数支持+-表示大于小于,ckMG表示数据单位
find [path] -size +10M #path下大于10M的文件
find [path] -size -1G #path下小于1G的文件时间戳
1
2
3
4find [path] -mtime -n +m #更改时间为m天前n天内的文件
find [path] -atime -n +m #访问时间
find [path] -ctime -n +m #创建时间
mtime参数后数字表示天, mmin参数后数字表示分钟文件属主
1
2
3
4find [path] -user <user_name> #path下属主为user_name的文件
find [path] -group <group_name> #path下组名为group_name的文件
find [path] -nouser #用户ID不存在的文件
find [path] -nogroup #组ID不存在的文件文件权限
1
2
3
4find [path] -perm 755 #权限设置为755的文件
find [path] -perm -u=r #文件属主有读权限的目录或文件
find [path] -perm -g=r #用户组有读权限的目录或文件
find [path] -perm -o=r #其它用户有读权限的目录或文件其他搜索参数
1
2
3
4find [path] <search_regular> [option]
-follow 追踪链接文件
-mount 不跨越文件系统mount点
-empty 空文件
示例
多筛选条件组合
1
2
3
4find [path] -name '*R' -a -mtime -1 #修改时间一天内的R脚本文件
-a | -and # 同时满足
-o | -or # 或
-not # 条件取反几个例子,来自微博@linux命令行精选网
1
2
3
4
5
6find . ! -name <NAME> -delete #用find删除文件时候排除特定文件
find . -type l -xtype l #查找失效的符号链接
find . -iname '*.jpg' | sed 's/.*/<img src="&">/' > gallery.html #生成html 相册
find . -size 0 -exec rm '{}' \; #清理空文件
find . -type d -exec mkdir -p $DESTDIR/{} \; #复制一个目录的结构,忽略文件
find . -type f -size +500M -exec ls -ls {} \; | sort -n #找出所有大于500M的文件
fasta 序列解析
fasta格式最常见,也最常用,也是分析中最简单的格式吧,以>开始后续字符表示序列名称,空格后对序列进行描述(可能不存在),换行即为序列信息,可包括换行。下面是一个fasta格式序列实例:
1 | >gi|187608668|ref|NM_001043364.2| Bombyx mori moricin (Mor), mRNA |
这个实例中序列名字为gi|187608668|ref|NM_001043364.2|,下面就直接贴上常用的fasta解析脚本:
python手动解析
1 | def parse_fasta(fa): |
使用HTSeq模块
1 | def parse_fasta_by_HTSeq(fa): |
当然还能使用biopython进行解析,我不太常用这一模块,就不写了
fasta输出格式化
1 | def format_fasta(seq, len=60): |
RefSeq 数据库 GFF 格式转换
最近一项目被RefSeq数据库中gff注释格式折磨死,流程总是不能正确识别,各种方法改了格式,勉强运行,后续步骤继续报错,想着以后肯定还有更多这种情况,自己对文件格式的理解,写了格式转换脚本。
GFF和GTF格式非常类似,都被用来基因结构注释,由9列表示不同意义的特征组成,多行共同注释一个基因的详细结构。然而两者又有很大差异,脚本处理起来也是不同。相比而言GTF处理起来简单些,第三列feature可能是gene, transcript, exon, CDS这少数几种情况,最重要的描述基因类型名称的第九列attr很统一,都会包括一般常用的gene_id, gene_biotype, transcript_id, transcript_biotype等特征。而GFF文件包括一个明确的层级关系,gene是transcript(存在mRNA,lncRNA,ncRNA,rRNA,tRNA等多种不同组织方式)的父节点,而transcript又是exon和CDS的父节点,特征层级不同也决定了第九列attr的注释信息不同,gene id, name等信息也是层次关系关联,文件处理较麻烦。
NCBI RefSeq数据库中GFF注释格式感觉更是复杂怪异,废话不多说,直接上代码,愚蠢方法手撕GFF
1 | #!/usr/bin/env python |
GFF格式包括gene, transcript, exon(CDS)三个层级,其中transcript进行上下关联最为重要,这里脚本以transcript作为键值进行数据处理,另外脚本还有一个假设就是gene, transcript对应ID在文件中唯一。